In medical device manufacturing, getting ISO 13485 certification ensures quality and regulatory compliance. This international standard sets the bar for quality management systems in the medical device industry. Companies seeking to enhance their processes, meet regulatory requirements, and gain a competitive edge often ask how to get ISO 13485 certification. It requires careful preparation and a series of well-defined steps.
If you’re ready to dive in, you can jump straight to the step-by-step guide below. This comprehensive guide will walk you through the ISO 13485 certification process. We’ll break down the key requirements and stages involved. Next, we will explore the importance of developing a robust quality management system, implementing effective risk management strategies, and ensuring proper training and competence. Also, this guide will discuss the role of measurement, analysis, and improvement in maintaining certification. Finally, we will provide insights on working with certification bodies.
By following this step-by-step process, you can lead your organization on the path to ISO 13485 certification with confidence and precision.
Understanding ISO 13485 Standard

History and Development
ISO 13485, the international standard for quality management systems in the medical device industry, has a rich history. It dates back to 1996 when the International Organization for Standardization (ISO) first published it to establish a comprehensive framework for designing and manufacturing medical devices. Since then, ISO 13485 has undergone significant revisions in 2003 and 2016. The current version, ISO 13485:2016, took effect in March 2016.
The standard’s evolution reflects the dynamic nature of the medical device industry and its regulatory landscape.
It responds to the latest quality management system practices, incorporates changes in technology and addresses the increasing regulatory requirements and expectations. This adaptability makes ISO 13485 a crucial tool for organizations involved in various aspects of the medical device lifecycle. These aspects include design, production, installation, and servicing.
Key Principles
ISO 13485 is built on several fundamental principles. These principles guide organizations in establishing and maintaining robust quality management systems. They are essential for ensuring the safety, effectiveness, and regulatory compliance of medical devices.
- Customer-Oriented Approach: The standard emphasizes understanding and meeting customer requirements and expectations. This includes patients, healthcare professionals, and regulatory bodies. By placing the customer at the center of the quality management system, you can deliver products that improve patient safety and satisfaction.
- Process Approach: ISO 13485 advocates for managing activities and resources as interrelated processes. This approach enables organizations to consistently deliver products that meet regulatory requirements and customer expectations. This is because you will understand and control these processes effectively.
- Risk-Based Decision Making: The standard promotes a systematic approach to risk. This includes identifying, analyzing, evaluating, and controlling risks throughout the product lifecycle. The process for addressing risks is best described in ISO 14971:2019 for risk management for medical devices. This principle helps organizations make informed decisions. That’s because you’ll be considering the potential impact of risks on product quality and patient safety.
- Continuous Improvement: ISO 13485 doesn’t require organizations to demonstrate continual improvement like ISO 9001. But, it does emphasize the need to maintain an effective quality management system. Organizations must consistently seek opportunities to improve their processes, products, and overall performance to meet evolving customer expectations and regulatory requirements.
- Documentation and Record-Keeping: The standard emphasizes the importance of maintaining documented information and records. This principle ensures effective planning, operation, control, and monitoring of processes and activities within the quality management system.
- Employee Involvement: ISO 13485 recognizes the value of employee participation in achieving quality objectives. It encourages organizations to involve employees at all levels. This fosters a culture of ownership and responsibility.
By adhering to these principles, organizations can establish a quality management system that meets the requirements of ISO 13485. But, this structure also serves as a foundation for regulatory compliance and customer satisfaction in the medical device industry.
Importance of ISO 13485 in the Medical Device Industry
ISO 13485 plays a crucial role in the medical device industry as an internationally recognized standard for quality management systems (QMS). This standard impacts regulatory compliance, quality assurance, and overall industry practices.
Regulatory Compliance
ISO 13485 is essential for medical device manufacturers to meet regulatory requirements across various markets. It’s not legally mandated for marketing medical devices in Europe. However, ISO 13485 certification helps adhere to EU Medical Device Regulation (MDR) requirements. The standard is harmonized with MDR and IVDR (In Vitro Diagnostic Regulation). This means companies that meet ISO 13485 requirements can presume to fulfill corresponding MDR and IVDR requirements.
For manufacturers looking to export their products, ISO 13485 certification is often necessary. In the United States, the FDA requires all medical devices sold to be aligned with Quality Management System Regulation (QMSR), which is aligned with ISO 13485 certification. This requirement extends to online marketplaces as well. For example, Amazon.com requires sellers to comply with the FDA requirements or to have a CE marking for the European market. Then, they can be listed as “Amazon Approved” in the medical device category. And ISO 13485 can help you align with these standards.
Quality Assurance
ISO 13485 provides a comprehensive framework for ensuring the quality and safety of medical devices throughout their lifecycle. The standard emphasizes:
- Risk Management: ISO 13485 focuses on risk-based decision-making processes by referencing ISO 14791. This helps manufacturers identify, analyze, and mitigate risks from the design stage to product use.
- Supplier Management: The standard sets rules for selecting and monitoring suppliers. This ensures that components and materials used in devices meet quality requirements.
- Documentation and Record-Keeping: ISO 13485 helps organize crucial documents and records. This facilitates audits and demonstrates compliance with regulatory requirements.
- Continuous Improvement: The standard encourages companies to regularly review their quality systems, identify issues, and implement improvements.
By implementing ISO 13485, manufacturers can:
- Reduce the risk of product recalls due to defects or malfunctioning parts;
- Avoid costly lawsuits from patients injured by defective products;
- Improve patient outcomes and increase customer satisfaction;
- Maintain a positive corporate image.
The QMS required by ISO 13485 is not a static set of documents. It’s a dynamic system that needs regular review and updates to ensure its continued effectiveness. This approach helps organizations maintain high standards of quality and safety in their medical devices. This ultimately benefits both manufacturers and end-users.
So, ISO 13485 serves as a cornerstone for quality management in the medical device industry. It provides a structured approach to regulatory compliance and quality assurance. Its implementation helps manufacturers navigate the complex landscape of medical device regulations. At the same time, it ensures the production of safe, effective, and high-quality devices.
How to Get ISO 13485 Certification – Step by Step
Obtaining ISO 13485 certification involves a systematic approach to implementing a quality management system (QMS) that meets the standard’s requirements. This process typically includes several key steps, each crucial for ensuring compliance and successful certification.

We can separate three main phases of pursuing certification:
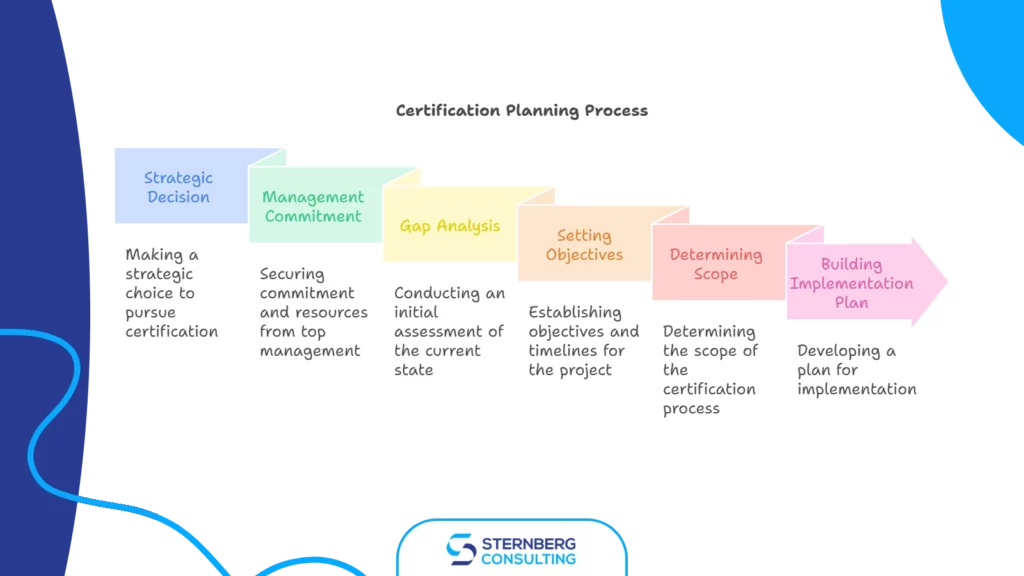
1) Planning
a) strategic decision to pursue certification, b) securing top management commitment and resources, c) gap analysis / initial assessment of current state, d) setting objectives and timelines, e) determining the scope of certification, f) building an implementation plan, g) creating a detailed project plan
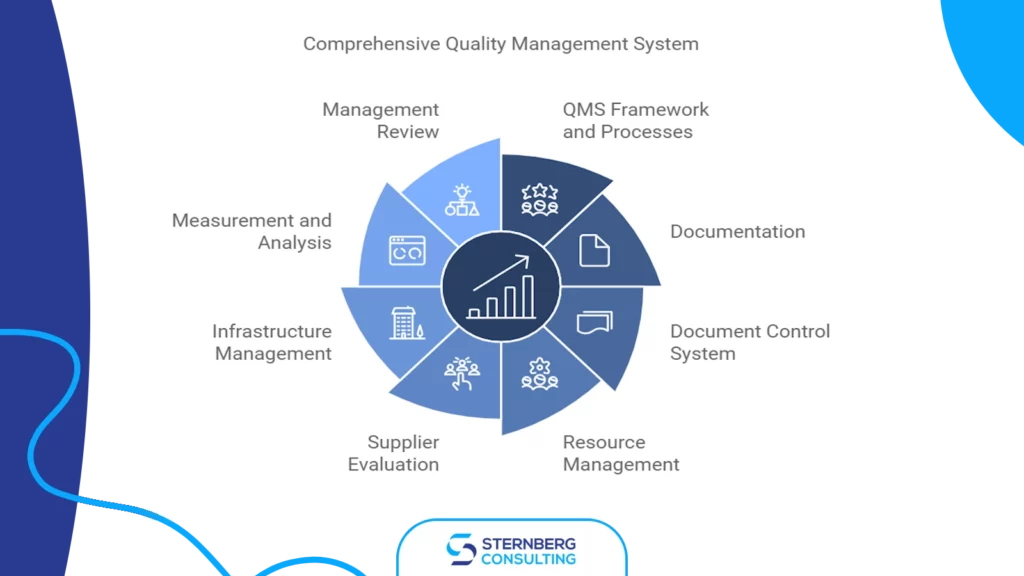
2) Developing and implementing a QMS
a) establishing QMS framework and processes, b) creating required documentation (policies, procedures) c) implementing document control system, d) setting up resource management processes, e) establishing supplier evaluation and management process, f) establishing and managing infrastructure (risk assessments, training programs, g) measurement, analysis, and internal auditing, h) management review process, i) establishing and implementing corrective action procedures
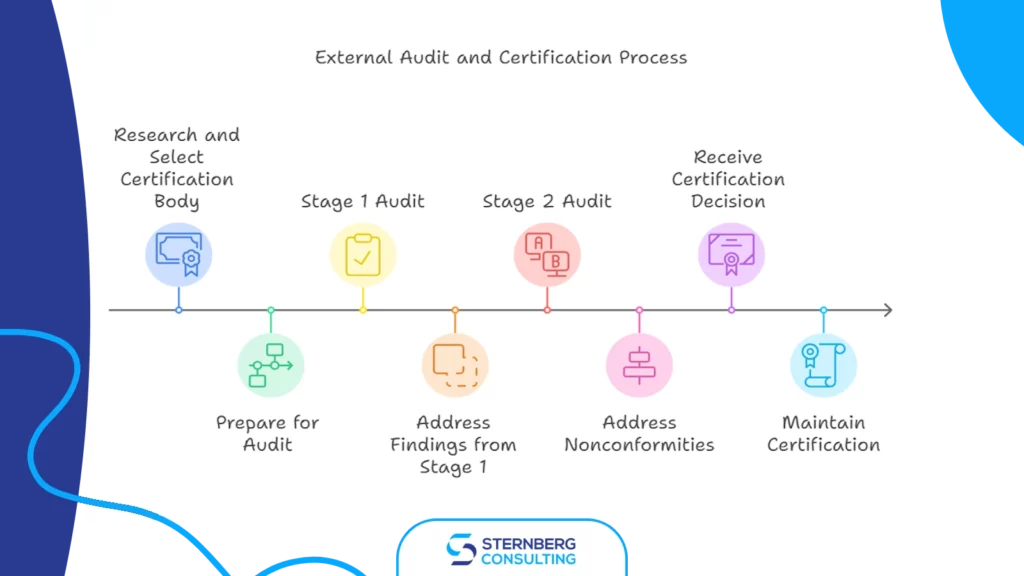
3) External audit/certification
a) researching and selecting an accredited certification body (like TUeV SUeD, Bureau Veritas, DNV, etc.), b) preparing for an audit, c) audit process (stage 1 audit (documentation review), addressing any findings from Stage 1, stage 2 audit (full system audit)) d) addressing nonconformities, e) receiving the certification decision, f) maintaining certification (surveillance audits, internal audits, continual improvement, recertification every 3 years)
Each of these phases includes multiple steps we’ll break down below.
Phase 1: Planning
Strategic Decision to Pursue Certification
The decision to pursue ISO 13485 certification is a strategic choice that reflects an organization’s commitment to quality, regulatory compliance, and customer satisfaction in the medical device industry. This decision should be based on:
- Market Demand: Understanding customer and regulatory expectations for certified quality management systems in the medical device sector.
- Competitive Advantage: Gaining a distinct edge in the marketplace by demonstrating a commitment to meeting international standards.
- Regulatory Compliance: Aligning with industry-specific regulations and ensuring products meet stringent requirements for safety and effectiveness.
- Organizational Growth Goals: Supporting the company’s long-term vision for growth and product innovation by building a robust Quality Management System (QMS).
Securing Top Management Commitment and Resources
Top management’s support is the cornerstone of a successful ISO 13485 implementation.

Their active involvement ensures the initiative is prioritized and adequately resourced. This stage includes:
- Leadership Advocacy: Executives must actively champion the certification process, signaling its importance to all levels of the organization.
- Resource Allocation: Ensuring sufficient funding, personnel, and time for the implementation and certification process.
- Alignment with Business Goals: Leadership should clearly connect the QMS objectives with organizational goals to highlight the strategic value of certification.
- Empowering a Team: Establishing a dedicated project team with authority and accountability for driving the certification process.
Initial Assessment of Current State with Gap Analysis
The journey to ISO 13485 certification begins with a detailed evaluation of the organization’s current practices to identify gaps and set the foundation for compliance. This phase focuses on:
- Understanding Current Practices:
- Review and document existing processes, procedures, and controls relevant to medical device manufacturing and quality management.
- Map out workflows to understand how they align—or diverge—from ISO 13485 requirements.
- Performing a Gap Analysis:
- Conduct a systematic comparison of current processes against the clauses of ISO 13485.
- Identify specific areas where existing practices fall short, such as incomplete documentation, lack of risk management processes, or gaps in supplier controls.
- Highlight high-priority areas that require immediate action to meet compliance standards.
- Prioritizing Improvements:
- Develop a clear list of actionable steps to close the identified gaps, categorized by urgency and impact on certification readiness.
- Focus on building the foundation for key processes required by ISO 13485, such as risk management, design controls, and product traceability.
Setting Objectives and Timelines
Well-defined objectives and realistic timelines create a clear roadmap for how to get ISO 13485 certification. Key steps include:
- Define Objectives: Develop specific, measurable, achievable, relevant, and time-bound (SMART) objectives for the certification journey, e.g.:
a) Achieve ISO 13485 certification by completing a gap analysis, implementing 100% of corrective actions, and passing the certification audit within 12 months.
b) Increase customer satisfaction scores by 10% within 6 months of ISO 13485 certification by resolving 90% of customer complaints within 2 weeks and improving quality assurance processes.
c) Reduce product defect rates from 5% to 2% within 9 months of ISO 13485 certification by implementing a structured quality management system and addressing quality issues through root cause analysis and corrective actions.
- Develop a Timeline: Map out the implementation phases, including:
- Training and awareness programs
- Process design and documentation
- Internal audits and corrective actions
- Certification audit readiness
- Monitor Progress: Include milestones and checkpoints to evaluate progress and address challenges promptly.
Determining the Scope of Certification
Clearly defining the scope of the QMS is critical to ensuring the certification process aligns with the organization’s operational and strategic priorities. Steps include:
- Identify Key Areas: Focus on processes directly related to medical devices, such as design, production, distribution, and support services.
- Understand Exclusions: Document any exclusions from the scope (if applicable) and justify them in alignment with ISO 13485 requirements.
- Align Scope with Objectives: Ensure the defined scope supports the organization’s quality objectives and compliance obligations.
- Communicate the Scope: Clearly communicate the QMS scope to stakeholders, including employees, customers, and regulatory bodies.
Building an Implementation Plan
After the initial assessment, organizations must develop a comprehensive implementation plan. This plan should include:
- Process Definition: Identify and document all processes relevant to the QMS, including:
- Mandatory procedures required by ISO 13485
- Company-specific processes that impact quality
- Process interactions and potential problem areas
- Documentation Design: Create necessary documentation, including:
- Quality manual outlining the QMS structure
- Quality policy stating the organization’s commitment to quality
- Procedures and work instructions for key processes
- Training Program: Develop and execute a training plan to:
- Inform employees about ISO 13485 implementation
- Explain individual responsibilities within the QMS
- Provide necessary skills and knowledge for effective implementation
- Implementation Schedule: Create a timeline with:
- Clearly defined, quantifiable objectives
- Realistic deadlines for each implementation phase
- Milestones for monitoring progress
- Resource Allocation: Assign internal auditors and teams to oversee various processes and implementation tasks.
- Continuous Improvement: Establish mechanisms for:
- Conducting internal audits to verify QMS effectiveness
- Implementing corrective and preventive actions (CAPA)
- Regularly reviewing and updating the QMS
A detailed implementation plan transforms the certification process into actionable steps, ensuring clarity and structure throughout the journey.
By following these steps, organizations can systematically approach the question of how to get ISO 13485 certification. This will ensure a robust QMS that meets regulatory requirements and enhances overall quality in medical device manufacturing.
Phase 2: Developing and Implementing Quality Management System
Developing a robust Quality Management System (QMS) is an important step in obtaining ISO 13485 certification. This process involves establishing a comprehensive framework that ensures consistent quality in medical device manufacturing. Two key components of an effective QMS are document control and resource management.
Establishing QMS Framework and Processes
Developing a Quality Management System (QMS) framework involves structuring processes and interactions to ensure consistent product quality, regulatory compliance, and customer satisfaction. Key steps include:
- Defining Core Processes: Identify all processes critical to medical device design, production, and delivery, such as design control, risk management, and product traceability.
- Mapping Process Interactions: Map out how these processes interact to form an integrated system, ensuring seamless communication and efficiency.
- Alignment with ISO 13485 Requirements: Structure the framework to meet all applicable clauses of the standard, including risk-based approaches and regulatory obligations.
- Customization: Tailor the QMS to the organization’s size, complexity, and product portfolio while maintaining compliance with ISO 13485.
Creating Required Documentation (Policies, Procedures)

A robust QMS requires comprehensive documentation to ensure consistency and traceability. This includes:
- Quality Policy and Objectives: A robust Quality Management System (QMS) requires comprehensive documentation to ensure consistency and traceability. Develop a quality policy demonstrating a commitment to meeting regulatory requirements and delivering safe, effective medical devices. The policy should be supported by measurable objectives aligned with organizational goals.
- Procedures and Work Instructions: Documented procedures must address essential processes like design and development, production control, nonconformity handling, purchasing, production and service provision, sterilization validation, identification, traceability, and product preservation, alongside monitoring and measuring equipment control. Specific procedures required by ISO 13485:2016 include validation of application software, document and record control, management reviews, competence and training, and work environment management.
In addition to these core processes, documented procedures for supporting activities such as risk management, clinical evaluation, market surveillance, and change management are essential.
- Quality Manual: Outline the structure and scope of the QMS, referencing relevant procedures and work instructions. A quality manual outlining the QMS structure, along with a systematic document control system, is fundamental to achieving regulatory standards and facilitating effective product realization.
- Document Control: Implement a document control system to ensure all documentation is reviewed, approved, and updated systematically.
- Additional Supporting Procedures Some procedures, while not explicitly required by ISO 13485, are vital for aligning with regulatory expectations, including MDR and IVDR, and ensuring operational success. Organizations should establish procedures for clinical investigations to validate the safety and performance of medical devices in real-world settings.
Procedures for conformity assessment ensure compliance with regulatory frameworks, while translation processes guarantee that documentation meets language requirements in different regions. Additionally, reprocessing procedures must be defined to manage the safe and effective reuse of medical devices where applicable.
- Continuous Improvement and Support Continuous improvement under ISO 13485 emphasizes proactive and systematic activities. Organizations must establish procedures for feedback mechanisms and complaint handling processes to capture and resolve customer issues effectively. Procedures for reporting to regulatory authorities ensure transparency and compliance. Regular audits and corrective action procedures drive operational improvements and address non-conformities.
Implementing a Document Control System
Document control serves as the backbone of an effective QMS. It encompasses all policies that ensure proper management of procedures, inputs, and outputs within a medical device company. A well-implemented document control system offers several benefits:
- Traceability: Enables quick and easy access to all documents and versions.
- Accountability: Creates greater levels of responsibility within the organization.
- Security: Keeps documents and information safe and protected.
- Compliance: Demonstrates adherence to regulatory requirements during audits.
To establish an effective document control system:
- Implement a review and approval process for new and modified documents.
- Ensure documents are clearly identifiable with unique titles and numbers.
- Make documents readily available to relevant employees.
- Control external documents necessary for the QMS.
- Properly manage obsolete documents by removing them from use and marking them as obsolete.
- Retain documents for at least the lifetime of the medical device or as specified by regulatory requirements.
Setting Up Resource Management Processes
Resource management is essential for maintaining an effective QMS. ISO 13485 requires organizations to ensure adequate resources are available to perform continuous work. This includes:
- Human Resources:
- Provide appropriate training and education to employees.
- Ensure employees have the necessary knowledge and skills for their roles.
- Document employee qualifications, including education, experience, and certifications.
- Evaluate the effectiveness of training periodically.
- Infrastructure:
- Maintain appropriate buildings, workspaces, and equipment.
- Implement proper maintenance procedures for facilities and equipment.
- Document requirements for infrastructure to achieve product conformity.
- Work Environment:
- Control environmental factors that may affect product quality.
- Implement processes to prevent contamination, especially for sterile medical devices.
- Document requirements for maintaining cleanliness during packaging.
By focusing on these key areas, medical device companies can develop a QMS that meets ISO 13485 requirements and supports the production of high-quality, safe devices. Remember, the strength of a medical device company is directly related to how effectively it manages documentation and resources. A well-structured QMS not only ensures compliance but also enhances overall organizational efficiency and product quality.
Establishing Supplier Evaluation and Management Process
Suppliers play a critical role in ensuring the quality and safety of medical devices. ISO 13485 requires organizations to implement a rigorous supplier management process, including:
- Supplier Evaluation:
- Establish criteria for selecting and evaluating suppliers, focusing on their ability to meet quality, regulatory, and delivery requirements.
- Perform initial evaluations through audits, questionnaires, or performance reviews.
- Ongoing Monitoring:
- Regularly assess supplier performance using metrics such as defect rates, delivery timelines, and compliance records.
- Supplier Agreements:
- Define quality requirements, roles, and responsibilities in formal agreements or contracts.
- Risk-Based Approach:
- Prioritize supplier evaluations based on the criticality of supplied components or services.
Establishing and Managing Infrastructure
Risk Management in ISO 13485
ISO 13485:2016 places significant emphasis on risk management throughout the quality management system (QMS) for medical devices. The standard requires organizations to implement a systematic approach to identifying, analyzing, evaluating, and controlling risks associated with the safety and performance of medical devices.

Risk Assessment
Risk assessment forms the foundation of effective risk management in ISO 13485. It involves identifying potential hazards, estimating the associated risks, and evaluating their significance. The standard defines risk as the combination of the probability of occurrence of harm and the severity of that harm. To conduct a comprehensive risk assessment, organizations should:
- Identify Hazards: Document potential risks in the Hazard Identification Document (HID), considering all aspects of the device’s design, production, storage, and usage.
- Analyze Risks: Evaluate each identified risk using two key parameters:
- Severity: Assess the potential impact if harm occurs
- Likelihood: Determine the probability of a harmful event occurring
- Evaluate Risks: Determine the significance of each risk based on the combination of severity and likelihood. This evaluation helps prioritize risks that require immediate attention and control measures.
- Document Findings: Record all risk assessment activities in the Risk Management File, which serves as a comprehensive repository of risk-related documentation for each product in the organization’s portfolio.
Risk Mitigation Strategies
Once risks have been assessed, ISO 13485 requires organizations to implement effective risk mitigation strategies. These strategies aim to reduce risks to acceptable levels and ensure the safety and performance of medical devices. Key aspects of risk mitigation include:
- Risk-Based Approach: Apply risk-based thinking to various aspects of the QMS, including:
- Design and development processes
- Training and competence management
- Supplier evaluation and selection
- Verification of purchased products and services
- Process Controls: Implement controls commensurate with the level of risk associated with each process. High-risk processes require more rigorous controls and monitoring.
- Validation and Verification: Conduct thorough validation and verification activities, particularly for:
- Software used in QMS processes
- Manufacturing processes
- Monitoring and measurement equipment
- Supplier Management: Apply a risk-based approach to control external providers, with more stringent criteria for suppliers of critical components or services.
- Continuous Monitoring: Establish processes for ongoing risk monitoring and evaluation, including:
- Collecting and analyzing post-production information
- Reviewing the Risk Management Plan before each Management Review
- Updating risk assessments based on new information or changes in processes
- Documentation: Maintain comprehensive documentation of all risk management activities, including:
- Risk Management Plan
- Risk analyses and evaluations
- Risk control measures
- Residual risk evaluations
By implementing these risk management strategies, organizations can enhance the safety and performance of their medical devices while ensuring compliance with ISO 13485 requirements. This systematic approach to risk management helps manufacturers identify potential issues early in the product lifecycle, implement effective controls, and continuously improve their QMS to meet evolving regulatory requirements and customer expectations.
Training and Competence Programs
ISO 13485:2016 places significant emphasis on the importance of training and competence in maintaining a robust quality management system for medical device organizations. The standard requires that all personnel involved in quality processes and specialized tasks possess the necessary skills and competency to execute their roles effectively.
Employee Awareness
Organizations must ensure that their employees are fully aware of the relevance and importance of their activities and how they contribute to the achievement of quality objectives.
This awareness serves as a cornerstone for maintaining high standards of quality throughout the organization. To achieve this, companies can implement various strategies:
- Regular communication: Use newsletters, group discussions, and bulletin boards to disseminate important information.
- Visual aids: Employ pictorial representations of processes, defects, and best practices using signboards throughout the workplace.
- Continuous reinforcement: Provide ongoing reminders and updates to keep employees informed about their role in maintaining quality.
By fostering a culture of awareness, organizations can minimize human errors, deviations, and non-conformances, ultimately contributing to improved product quality and patient safety.
Skill Development

To meet ISO 13485 requirements, organizations must implement a comprehensive approach to skill development:
- Competency mapping: Document the process of establishing competency for each role, identifying skill gaps, and determining relevant training needs.
- Training programs: Provide appropriate education, training, and other learning activities to develop the required knowledge and skills. The frequency and intensity of training should be proportionate to the risk associated with the work and its impact on quality.
- Effectiveness assessment: Evaluate the effectiveness of training programs through various methods:
- Written tests and quizzes
- Monitoring process efficiency and product quality improvements
- Regular work performance evaluations
- Record-keeping: Maintain detailed records of all training programs, including employee files, certificates, and trainer competency documentation.
- Continuous improvement: Conduct periodic training to maintain a high level of competency within the organization.
- Risk-based approach: Consider training as a risk mitigation tool, focusing more resources on high-risk areas to minimize potential quality issues.
- End-user training: When necessary, provide training to end-users of medical devices to ensure safe and proper use according to the intended purpose.
Organizations should view training and competence as critical components of their quality management system. By investing in employee development and maintaining a skilled workforce, medical device manufacturers can enhance product quality, ensure regulatory compliance, and ultimately contribute to improved patient outcomes.
To effectively manage training and competence, organizations should establish a documented process that includes:
- Determining the necessary competencies for each role
- Providing appropriate training or taking other actions to achieve competence
- Evaluating the effectiveness of training activities
- Ensuring employee awareness of their role in quality objectives
- Maintaining comprehensive records of education, training, skills, and experience
By adhering to these principles and implementing a robust training and competence program, medical device organizations can meet ISO 13485 requirements while fostering a culture of continuous improvement and excellence in quality management.
Measurement, Analysis, and Internal Auditing
ISO 13485 emphasizes the importance of measurement, analysis, and improvement in maintaining an effective quality management system (QMS) for medical device organizations. This section of the standard requires companies to collect and analyze data from various processes and activities to identify trends, patterns, and opportunities for enhancement.
The foundation of an effective measurement and analysis process lies in robust data collection. ISO 13485 mandates that organizations gather information from multiple sources to verify the continuing suitability and effectiveness of their QMS. Key aspects of data collection include:
- Process Monitoring: Collect data from various QMS processes, including design and development, production, and customer feedback.
- Resource Management: Gather information on human resources, work environment, and infrastructure to ensure they meet quality requirements.
- Product Quality Planning: Collect data on product specifications, manufacturing processes, and quality control measures.
- Risk Management: Compile information on risk control performances and their effectiveness.
- Validation and Verification: Gather data on the results of validation and verification activities throughout the product lifecycle.
Organizations should establish a systematic approach to data collection, ensuring that the information gathered is relevant, accurate, and timely. This data serves as the input for analysis and improvement activities.

So, after all your procedures, policies, controls, and measurements have been in place for a while, you will have some data. And it would make sense to check the effectiveness of what you’ve implemented. That will help you prepare for certification. Besides, ISO 13485 emphasizes regular internal audits as a mechanism for maintaining QMS effectiveness and identifying areas for improvement. This process aligns closely with measurement, analysis, and improvement requirements. Key steps include:
- Audit Planning:
- Develop an audit schedule covering all QMS processes within a defined timeframe, ensuring a risk-based approach to prioritize critical areas.
- Training Auditors:
- Train internal auditors on ISO 13485 requirements, auditing techniques, and objectivity to ensure comprehensive evaluations.
- Executing Audits:
- Use standardized checklists and gather evidence through interviews, document reviews, and process observations.
- Evaluate compliance with documented procedures and ISO 13485 requirements.
- Audit Reporting:
- Document findings, highlighting nonconformities, observations, and opportunities for improvement.
- Communicate results to process owners and management.
- Corrective Actions:
- Ensure timely resolution of identified nonconformities through a formal corrective action process.
- Follow-Up:
- Verify the effectiveness of corrective actions during subsequent audits or reviews.
Internal audits form a feedback loop that ensures continuous improvement and prepares the organization for external certification audits.
Management Review Process
Your new QMS has been functioning for a while now. You have:
- Collected operational data
- Completed internal audits
- Analyzed performance indicators across business processes
Congratulations, you’re almost there. Now you’re ready for a high-level assessment by top management. This assessment is known as management review. It will evaluate the overall effectiveness of the management system and make strategic decisions for continual improvement.
The process includes:
- Scheduled Reviews: Conduct regular management reviews, typically on a quarterly or annual basis.
- Review Inputs: Include internal audit findings, process performance, customer feedback, supplier performance, and non-conformities.
- Analysis and Decision-Making:
- Assess the adequacy of resources, risks, and opportunities for improvement.
- Define corrective actions or strategic initiatives to address identified issues.
- Outputs: Document review outcomes, including decisions on policy updates, process changes, or resource allocations.
Establishing and Implementing Corrective Action Procedures
While quality issues are inevitable in complex manufacturing environments, the true measure of excellence lies in how organizations respond and learn. Corrective actions procedures create the structured pathway from problem identification to permanent resolution. So, they play a crucial role in addressing identified issues and preventing their recurrence. ISO 13485 requires organizations to implement a robust Corrective and Preventive Action (CAPA) process. Key elements of an effective corrective action process include:
- Root Cause Analysis: Conduct thorough investigations to identify the underlying causes of nonconformities or potential issues.
- Action Planning: Develop and document appropriate corrective actions based on the root cause analysis.
- Implementation: Execute the planned corrective actions promptly and effectively.
- Effectiveness Monitoring: Track and evaluate the impact of implemented corrective actions to ensure they address the identified issues without introducing new problems.
- Documentation: Maintain comprehensive records of the entire corrective action process, including root cause analysis, action plans, and effectiveness evaluations.
Organizations should establish a cross-functional team, often referred to as a Management Review Board (MRB), to review and discuss issues that may require corrective actions. This team typically includes representatives from quality, regulatory, operations, and engineering departments.
To enhance the effectiveness of measurement, analysis, and improvement processes, organizations should consider the following best practices:
- Set Clear Goals: Establish measurable objectives for each QMS process to facilitate performance evaluation.
- Utilize Statistical Techniques: Apply appropriate statistical methods to analyze data and identify trends or patterns.
- Implement a Robust CAPA System: Develop a well-defined CAPA process that addresses both corrective and preventive actions.
- Leverage Technology: Utilize quality management software to streamline data collection, analysis, and reporting processes.
- Foster a Culture of Continuous Improvement: Encourage employees at all levels to contribute to the identification and resolution of quality issues.
By implementing these practices and adhering to ISO 13485 requirements, medical device organizations can establish a robust system for measurement, analysis, and improvement. This approach not only ensures compliance with regulatory standards but also drives continuous enhancement of product quality and patient safety.
Phase 3: External Audit/Certification
Researching and Selecting an Accredited Certification Body
Organizations seeking ISO 13485 certification must choose a reputable certification body to conduct the external audit. While ISO 13485 itself does not mandate selection of an accredited certification body, it is highly recommended to choose one that complies with ISO 17021. Such accredited bodies undergo independent assessments, ensuring their competence and impartiality. In contrast, for products that fall under the scope of the European Medical Device Regulation (MDR) or In Vitro Diagnostic Regulation (IVDR), the involvement of a formally designated Notified Body is a legal requirement. These Notified Bodies must meet strict regulatory criteria to ensure a compliant conformity assessment.
When selecting an auditor, consider the following factors:
- Accreditation status
- Experience in the medical device industry
- Reputation and track record
- Availability and scheduling flexibility
- Cost and value for services provided
Preparing for an Audit
Preparing for an ISO 13485 audit requires careful planning and organization. To prepare effectively for an ISO 13485 audit, organizations should:
- Set clear goals and allocate sufficient time to meet standard requirements.
- Inform all employees about the audit scope and schedule.
- Ensure employees understand quality objectives and their roles in achieving them.
- Provide proper training to all employees on their tasks and responsibilities.
- Update document and record lists, ensuring all documentation is current and approved.
- Verify that processes and procedures are followed consistently by all employees.
- Maintain a clean and organized facility to prevent overlooking nonconformances.
- Conduct thorough internal audits to identify and address potential issues.
- Perform management reviews following internal audits to address findings and implement corrective actions.
- Consider a pre-assessment audit to identify areas for improvement before the official certification audit.
Organizations should emphasize that audits are not tests but opportunities to demonstrate how employees access information and follow procedures. Proper preparation helps ensure a smooth audit process and increases the likelihood of successful ISO 13485 certification.
Audit Process
The external audit process for ISO 13485 certification typically involves two stages, each critical for verifying compliance and readiness.
- Stage 1 Audit: Documentation Review
a)The certification body conducts an initial review of your Quality Management System (QMS) documentation.
b) This includes assessing the quality manual, policies, procedures, and other key documents to ensure they meet ISO 13485 requirements.
c) Auditors evaluate whether your organization is prepared for the Stage 2 audit by examining the scope, objectives, and implementation progress. They provide an audit report detailing areas of compliance and non-compliance, allowing the organization to implement necessary corrective actions.
- Addressing Stage 1 Findings
a) Any nonconformities or gaps identified during the Stage 1 audit must be addressed before moving to the next phase.
b) This involves corrective actions, such as updating documentation, refining processes, or providing additional evidence of compliance.
c) Once resolved, the certification body will confirm readiness for the Stage 2 audit.
- Stage 2 Audit: Full System Audit
a) The certification body evaluates the implementation and effectiveness of the QMS.
b) Auditors assess compliance across all processes, including design controls, risk management, supplier management, and production activities.
c) Evidence is gathered through interviews, process observations, and records review.
d) The audit concludes with a detailed report highlighting any findings, including major or minor nonconformities that require action.
Addressing Nonconformities
Nonconformities identified during the audit process must be resolved to achieve certification. The steps include:
- Classification of Nonconformities:
a) Major Nonconformities: Issues that significantly impact compliance or product safety, requiring immediate attention.
b) Minor Nonconformities: Less critical deviations that do not compromise overall compliance but must still be addressed. - Corrective Action Plan:
a) Develop a detailed plan to address each nonconformity, including root cause analysis, corrective actions, and timelines.
b) Submit the plan to the certification body for approval. - Implementation and Verification:
a) Implement the corrective actions and document the changes made.
b) Provide evidence to the certification body to verify the issues have been resolved. - Re-Audit (if necessary):
a) For major nonconformities, the certification body may conduct a follow-up audit to confirm compliance.
Receiving the Certification Decision
After addressing all findings from the audit, the certification body will make a decision on ISO 13485 certification.
- Final Evaluation:
a) This comprehensive review ensures that the QMS meets ISO 13485 requirements and operates effectively.
b) The certification body reviews the audit findings, corrective actions, and any additional evidence provided.
- Certification Issuance:
a) If compliance is confirmed, the organization receives an ISO 13485 certificate, valid for three years.
b) The certificate includes the scope of certification, demonstrating the organization’s commitment to quality and regulatory compliance.
- Next Steps:
a) The certification decision marks the beginning of ongoing maintenance to uphold compliance through surveillance audits and continual improvement.

Maintaining Certification
Achieving ISO 13485 certification is a continuous process that requires regular monitoring and improvement to sustain compliance.
- Surveillance Audits:
- Conducted annually by the certification body to ensure ongoing compliance with ISO 13485 requirements.
- Focus areas typically include key processes, corrective actions from previous audits, and any changes to the QMS or operations.
- Internal Audits:
- Organizations must perform regular internal audits to monitor the effectiveness of their QMS.
- Internal audits help identify potential nonconformities and opportunities for improvement before external audits.
- Continual Improvement:
- Use data from audits, customer feedback, and process performance to identify areas for enhancement.
- Implement risk-based thinking to address emerging challenges and drive innovation.
- Recertification:
- Every three years, organizations undergo a comprehensive re-audit to renew their certification.
- This process evaluates the entire QMS and ensures it remains compliant and effective.
Maintaining certification demonstrates an ongoing commitment to quality, regulatory compliance, and the delivery of safe, effective medical devices.
Conclusion
How to get ISO 13485 certification is a common question for medical device manufacturers. They need to ensure quality, regulatory compliance, and competitiveness in the industry. The journey to certification involves a systematic approach, including thorough preparation, implementation of a robust quality management system, and effective risk management strategies. Organizations must focus on developing comprehensive documentation, fostering employee awareness and competence, and establishing processes for continuous measurement, analysis, and improvement.
The path to ISO 13485 certification requires commitment, attention to detail, and a culture of quality throughout the organization. By following the steps outlined in this guide, companies can navigate the certification process with confidence and precision, ultimately enhancing their processes and meeting regulatory requirements. Are you ready to improve your quality management? Contact us now and let’s discuss how we can work together to achieve your ISO certification goals.
FAQs
What is the cost to become ISO 13485 certified?
The cost of obtaining ISO 13485 certification can vary widely depending on various factors such as the size of the company, the complexity of the medical devices manufactured, and the specific requirements of the quality management system.
Which organizations are authorized to issue ISO 13485 certifications?
ISO 13485 certificates are issued by certification or registration bodies, also known as Registrars or CBs (or notified bodies). These bodies are independent of the International Organization for Standardization (ISO) and must be accredited by a member of the International Accreditation Forum (IAF) to gain international recognition. This accreditation is crucial for businesses operating globally.
What documents are necessary to obtain ISO 13485 certification?
To achieve ISO 13485 certification, you need to prepare several documents that integrate elements of ISO 9001’s previous version. These include:
- Quality manual
- Quality policies and procedures
- Software validation process
- Medical device file
- Record of management participation
- Employee records
- Infrastructure and maintenance records
- Pollution control measures
What documents are necessary to obtain ISO 13485 certification?
To obtain ISO 13485 certification, an organization must implement a documented quality management system that demonstrates a commitment to regulatory compliance, product safety, and effectiveness. This begins with a quality policy that aligns with organizational objectives and includes measurable targets. The required documentation extends to procedures for design and development, production control, risk management, nonconformity handling, and validation of application software. It also encompasses document and record control, regular management reviews, competence and training measures, and maintaining a controlled work environment.
Such procedures guide all aspects of product realization, from design, development, purchasing, production, and service provision, to sterilization validation, identification, traceability, product preservation, and the monitoring and measurement of equipment. Continuous improvement is assured through documented methods for collecting feedback, handling complaints, reporting to regulatory authorities, conducting audits, and implementing corrective actions. Supporting activities include risk management, clinical evaluation, market surveillance, and change management, ensuring that the system evolves with regulatory requirements.
While not always explicitly required by ISO 13485, additional procedures help align with broader regulations, including MDR and IVDR. These address clinical investigations in real-world conditions, conformity assessment, appropriate translation of documentation for different regions, and reprocessing methods for safely reusing medical devices. All these procedures are anchored by a quality manual that defines the structure of the entire system and supported by a robust document control framework that ensures consistency, transparency, and prompt regulatory readiness.
How much does ISO 13485 training cost?
The cost of training for ISO 13485 varies depending on the course:
- ISO I3485 Internal Auditor Training course costs approximately $1400.
- ISO 13485 Lead Auditor Training course costs $2100.