Wandel ist unvermeidlich, aber in der Medizintechnikbranche ist er auch eine Chance – zur Innovation, zur Verfeinerung und zum Aufbau von Vertrauen. Die Implementierung des Änderungskontrollmanagements in ISO 13485-Systemen verwandelt diesen ständigen Fluss in eine gut orchestrierte Symphonie aus Qualität, Sicherheit und Compliance. Ob es um die Aktualisierung eines Designs, die Optimierung eines Prozesses oder die Zusammenarbeit mit einem neuen Lieferanten geht – ein robustes Änderungsmanagement stellt sicher, dass jede Anpassung zu einem strategischen Vorteil wird. Indem Hersteller den Wandel als Werkzeug für Wachstum begreifen, erfüllen sie nicht nur regulatorische Anforderungen, sondern festigen auch ihr Engagement für Exzellenz und Zuverlässigkeit.
Wie geht man dabei vor? Das ist genau das Thema des heutigen Blogs, also tauchen wir direkt ein!
Dokumentenlenkung im Änderungsmanagement
Die Dokumentenlenkung spielt eine entscheidende Rolle bei der effektiven Verwaltung von Änderungen innerhalb der Medizintechnikbranche. Sie stellt sicher, dass alle Modifikationen an Produkten, Managementprozessen und Dokumentationen systematisch implementiert werden, wobei die Konformität mit der ISO 13485 und den regulatorischen Anforderungen gewahrt bleibt.
Revisionskontrolle
Ein robustes Revisionskontrollsystem ist unerlässlich für die Verwaltung von Dokumentenversionen, Aktualisierungen und Änderungen. Organisationen müssen eine zuverlässige und standardisierte Richtlinie für das Dokumentenmanagement einführen.
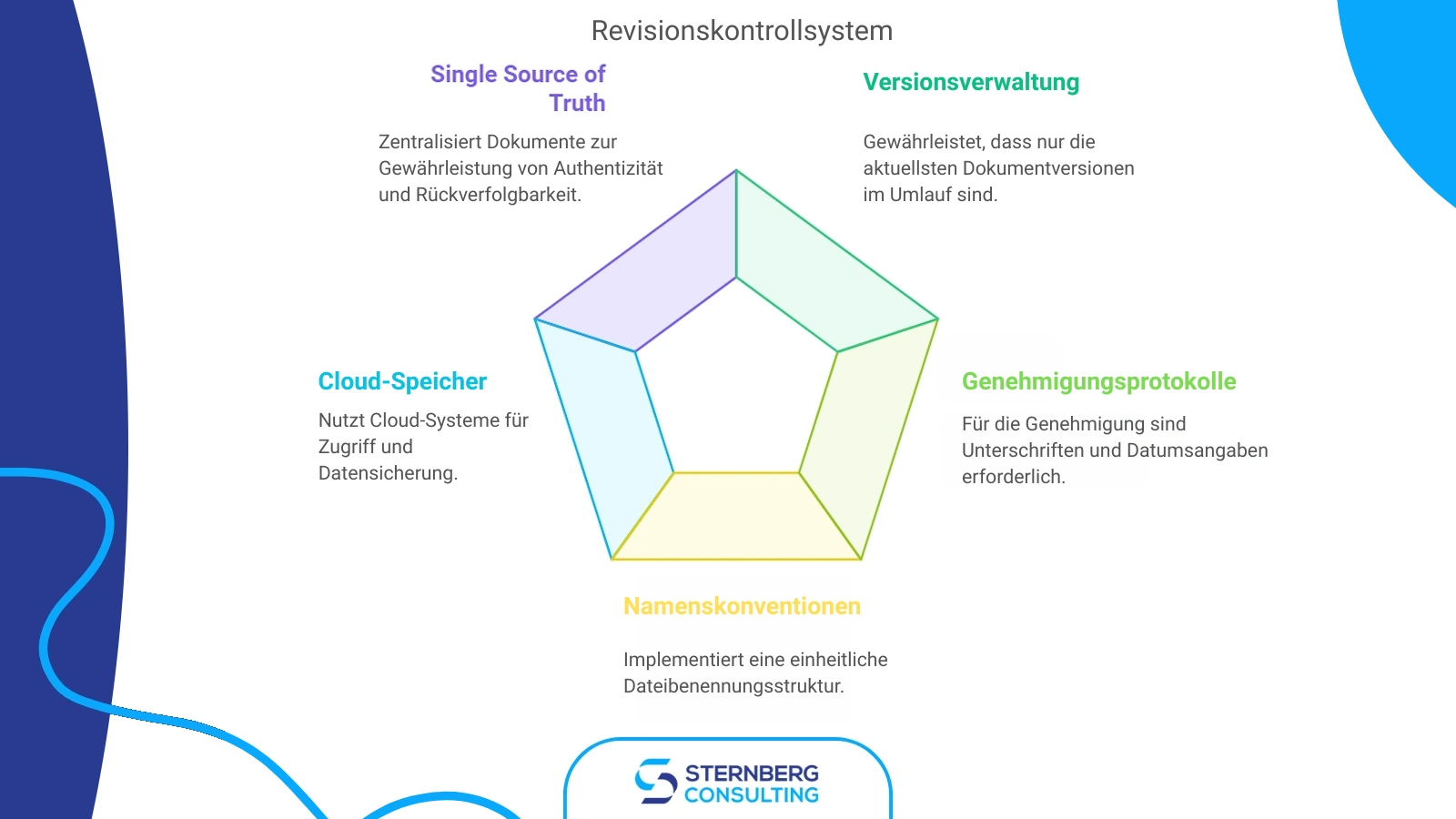
Wichtige Aspekte der Revisionskontrolle umfassen:
-
Versionsverwaltung: Stellen Sie sicher, dass nur die neuesten, aktuellen Versionen von Dokumenten im Umlauf sind und an den entsprechenden Standorten zur Verfügung stehen.
-
Freigabeprotokolle: Alle Genehmigungen zur Dokumentenlenkung müssen die Unterschriften der genehmigenden Person(en) und das Datum der Genehmigung enthalten. Änderungen müssen von den Personen überprüft und genehmigt werden, die ursprünglich an der ersten Prüfung und Genehmigung beteiligt waren.
-
Standardisierte Benennungskonventionen: Implementieren Sie eine konsistente Dateibenennungsstruktur, um Dokumenteninhalte und deren Beziehungen zu anderen Dateien zu beschreiben.
-
Nutzung von Cloud-Speichern: Speichern Sie Dateien auf cloudbasierten Systemen, um die Zugänglichkeit und Datensicherung im Falle von Serverausfällen zu gewährleisten.
-
Single Source of Truth (Einzige Quelle der Wahrheit): Zentralisieren Sie alle Dokumente in einem einzigen Repository, um Authentizität und Rückverfolgbarkeit während des gesamten Produktlebenszyklus zu gewährleisten.
Profi-Tipp: Um den Aufwand der manuellen Revisionsverwaltung zu eliminieren, investieren Sie in ein zuverlässiges Dokumentenmanagementsystem (DMS). Ein gutes DMS automatisiert die Versionskontrolle, rationalisiert Genehmigungs-Workflows und zentralisiert die Speicherung, wodurch sichergestellt wird, dass nur die neuesten, genehmigten Versionen zugänglich sind. Achten Sie auf Funktionen wie cloudbasierten Zugriff, digitale Signaturen und anpassbare Benennungskonventionen. Dies stellt nicht nur die Konformität mit ISO 13485 sicher, sondern spart auch Zeit, reduziert Fehler und gibt Ihnen Sicherheit bei Audits und im täglichen Betrieb.
Kommunikation von Änderungen
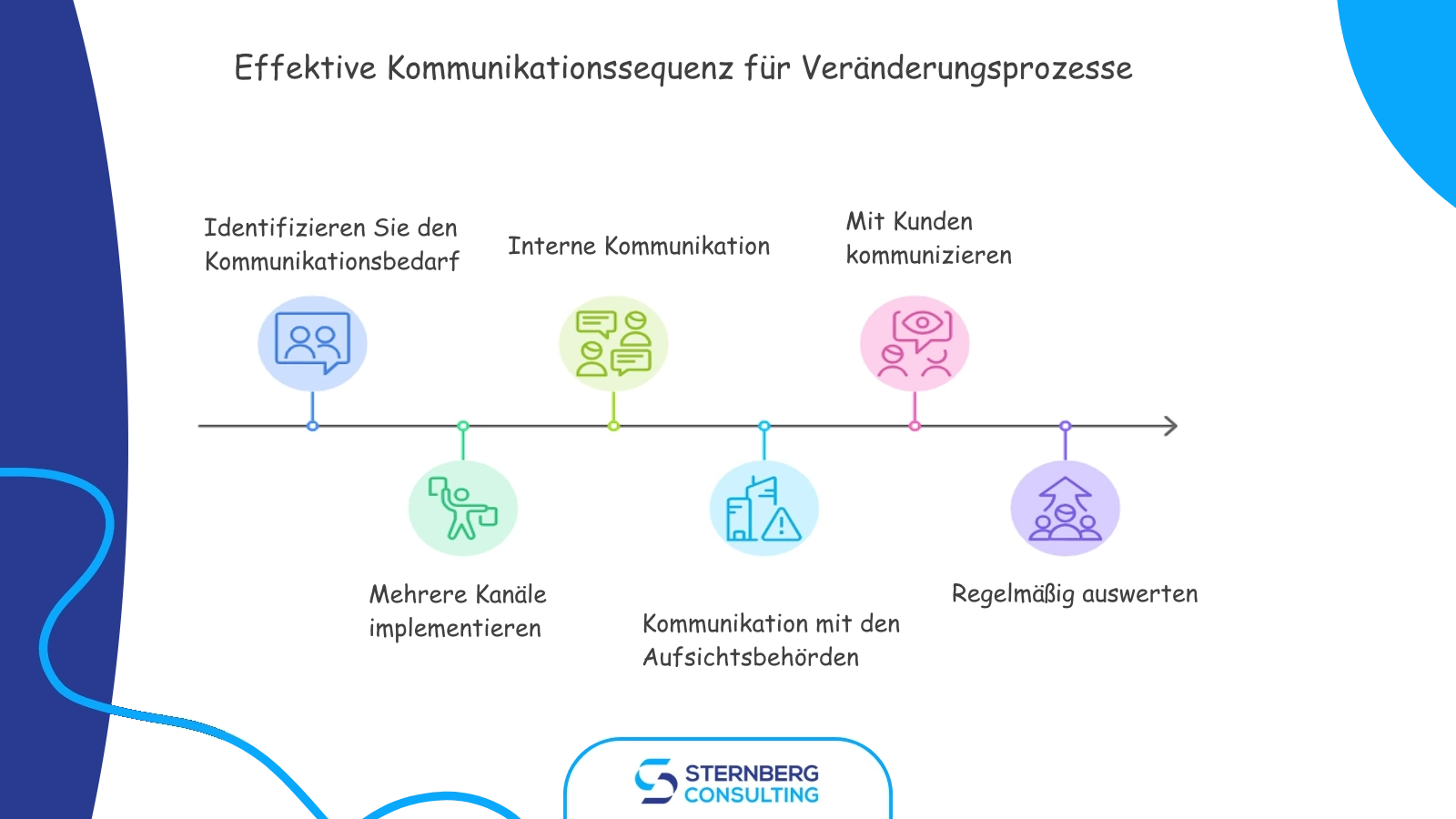
Effektive Kommunikation ist die Säule für ein erfolgreiches Änderungsmanagement. Mangelhafte Kommunikationspraktiken können zu operativen Verlusten und Compliance-Problemen führen. Um eine effiziente Kommunikation von Änderungen zu gewährleisten:
-
Implementieren Sie mehrere Kommunikationskanäle: Nutzen Sie verschiedene Methoden wie E-Mail, Telefon und formelle Benachrichtigungen, um alle Stakeholder effektiv zu erreichen.
-
Kommunizieren Sie intern: Fördern Sie eine kollaborative Lernumgebung, indem Sie Mitarbeiter ermutigen, Erfahrungen, Erkenntnisse und Tipps im Zusammenhang mit Dokumentenmanagement und Änderungskontrolle zu teilen.
-
Kommunizieren Sie mit Aufsichtsbehörden: Beauftragen Sie qualifizierte Vertreter damit, die Behörden über bedeutende Reklamationen, meldepflichtige Ereignisse, Sicherheitsmitteilungen im Feld (FSN) und Aktualisierungen der technischen Dokumentation zu informieren, welche die Produktsicherheit, Qualität oder Wirksamkeit beeinträchtigen könnten.
-
Kommunizieren Sie mit Kunden: Legen Sie effektive Regelungen für die Einholung von Kundenfeedback und die Kommunikation über Produktinformationen, Anfragen, Verträge und Rückmeldungen fest und setzen Sie diese um.
-
Regelmäßige Bewertung: Bewerten Sie regelmäßig die bestehenden Kommunikationswerkzeuge und nehmen Sie Verbesserungen vor, um einen robusten Kommunikationsprozess zu gewährleisten.
Schulung zu Aktualisierungen
Die Bereitstellung gründlicher Schulungen zu allen Aktualisierungen oder Änderungen am Dokumentenmanagementsystem ist für die Aufrechterhaltung der Compliance und der operativen Effizienz unerlässlich. Um diese Schulungen wirklich effektiv zu gestalten, berücksichtigen Sie Folgendes:
-
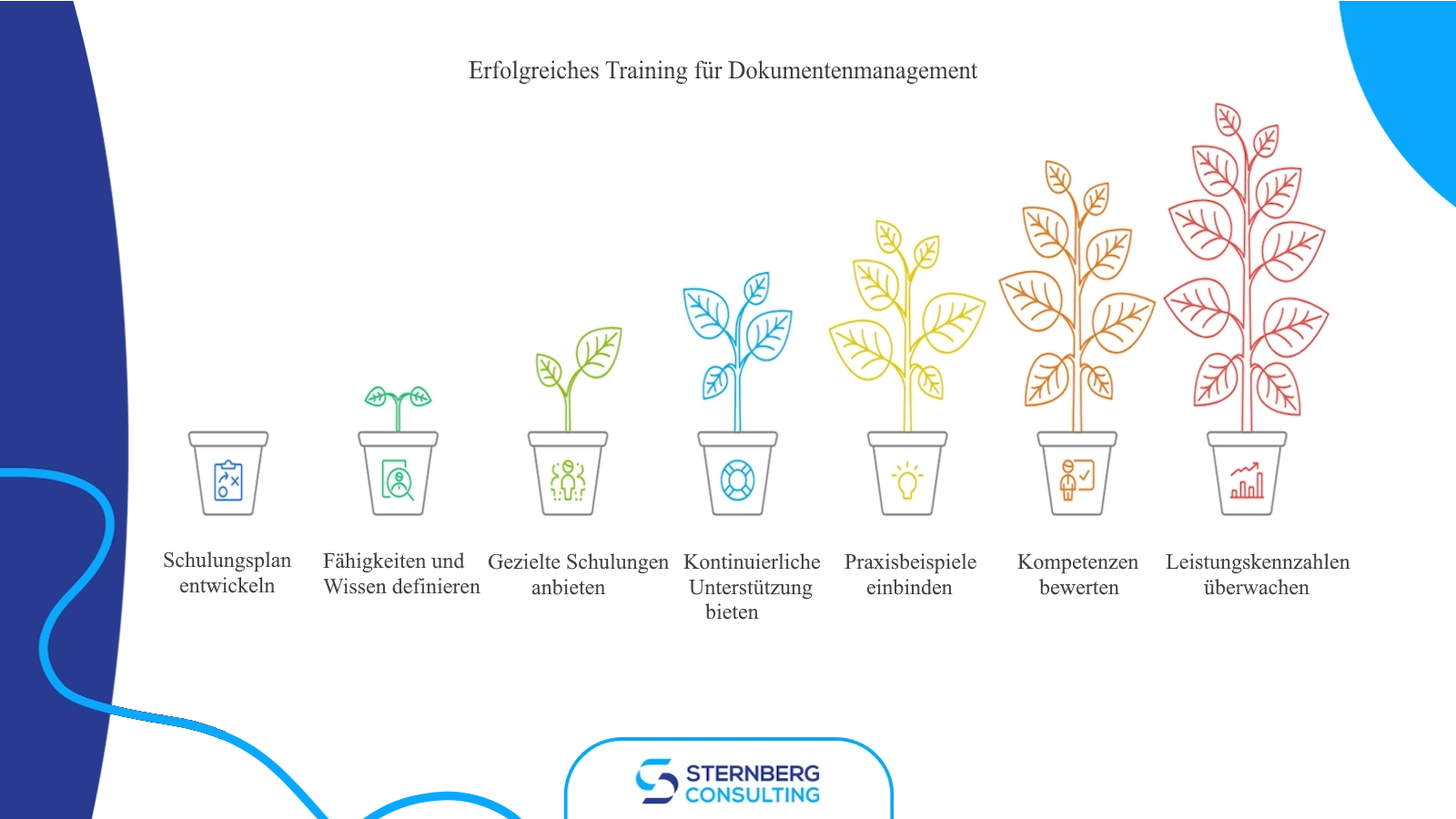
Entwickeln Sie einen detaillierten Schulungsplan: Skizzieren Sie Ziele, Zeitplan und Schulungsmethoden für die Ausbildung der Mitarbeiter in Bezug auf Dokumentenmanagementsysteme.
-
Definieren Sie spezifische Fähigkeiten und Kenntnisse: Identifizieren Sie klar die Kompetenzen, die Mitarbeiter durch die Schulung erwerben sollten, wie etwa das Verständnis der DMS-Struktur und die Nutzung fortgeschrittener Suchfunktionen.
-
Bieten Sie gezielte Schulungen an: Passen Sie das Schulungsprogramm an die spezifischen Bedürfnisse und Verantwortlichkeiten der verschiedenen Mitarbeiterrollen innerhalb der Organisation an.
-
Bieten Sie kontinuierliche Unterstützung an: Erkennen Sie an, dass Lernen ein fortlaufender Prozess ist, und bieten Sie Auffrischungskurse, Fortgeschrittenenschulungen und regelmäßige Updates zu neuen Funktionen und Best Practices an.
-
Integrieren Sie praxisnahe Beispiele: Demonstrieren Sie praktische Anwendungen des Dokumentenmanagementsystems anhand spezifischer, für die Organisation relevanter Anwendungsfälle.
-
Bewerten Sie die Kompetenz der Mitarbeiter: Führen Sie während und nach den Schulungen Bewertungen und Quizfragen durch, um das Verständnis der Mitarbeiter zu messen und Verbesserungsbereiche zu identifizieren. Diese Bewertungen dienen später auch als Nachweis der Konformität.
-
Überwachen Sie Leistungskennzahlen: Beobachten Sie, wie Mitarbeiter das Dokumentenmanagementsystem nutzen, um die Leistung zu messen und Bereiche zu identifizieren, in denen Verbesserungen möglich sind. Nutzen Sie diese Informationen, um gezielte Schulungen zu erstellen, die den Mitarbeitern helfen, effizienter und effektiver zu arbeiten.
Durch die Implementierung dieser Strategien für Revisionskontrolle, Änderungskommunikation und Schulung zu Aktualisierungen können Organisationen eine effektive Dokumentenlenkung im Änderungsmanagement sicherstellen.
Profi-Tipp: Obwohl es noch nicht weit verbreitet ist, kann die Implementierung eines Learning Management Systems (LMS) ein entscheidender Vorteil für Schulungen zu Dokumentenmanagementsystemen sein. Ein LMS ermöglicht es Ihnen, maßgeschneiderte, rollenspezifische Schulungsmodule zu erstellen, auf die Mitarbeiter nach Belieben zugreifen können. Die Einbindung interaktiver Elemente wie Video-Tutorials, Quizfragen und realer Szenarien macht den Lernprozess ansprechend und einprägsam. Darüber hinaus verfolgt ein LMS den Schulungsfortschritt und den Abschluss, was dokumentierte Konformitätsnachweise liefert – ein wertvolles Gut bei Audits. Durch die Kombination eines LMS mit fortlaufender Unterstützung, wie Auffrischungskursen und speziellen Hilfe-Kanälen, können Sie sicherstellen, dass die Mitarbeiter im Umgang mit aktualisierten Systemen sicher und kompetent bleiben, was Ihre Organisation in Bezug auf die operative Effizienz von anderen abhebt.
Implementing Change Control Management in ISO 13485: Verwaltung von Prozessänderungen
Das Management von Prozessänderungen ist ein weiterer Aspekt der Aufrechterhaltung von Qualität und Compliance in der Herstellung von Medizinprodukten. Es beinhaltet den systematischen Umgang mit Modifikationen an Herstellungsprozessen, Qualitätskontrollverfahren und Ausrüstung. Dieser Ansatz stellt sicher, dass Änderungen kontrolliert implementiert werden, wobei Produktsicherheit und Wirksamkeit gewahrt bleiben, während gleichzeitig die regulatorischen Anforderungen eingehalten werden.
Änderungen am Herstellungsprozess
Hersteller von Medizinprodukten müssen ihre Herstellungsprozesse oft aufgrund verschiedener Faktoren modifizieren. Dies können Änderungen in der Lieferkette, Initiativen zur fortlaufenden Verbesserung oder technologische Fortschritte sein. Diese Änderungen können die Sicherheit und Wirksamkeit des Produkts erheblich beeinflussen. Um solche Modifikationen effektiv zu verwalten, sollten Hersteller:
-
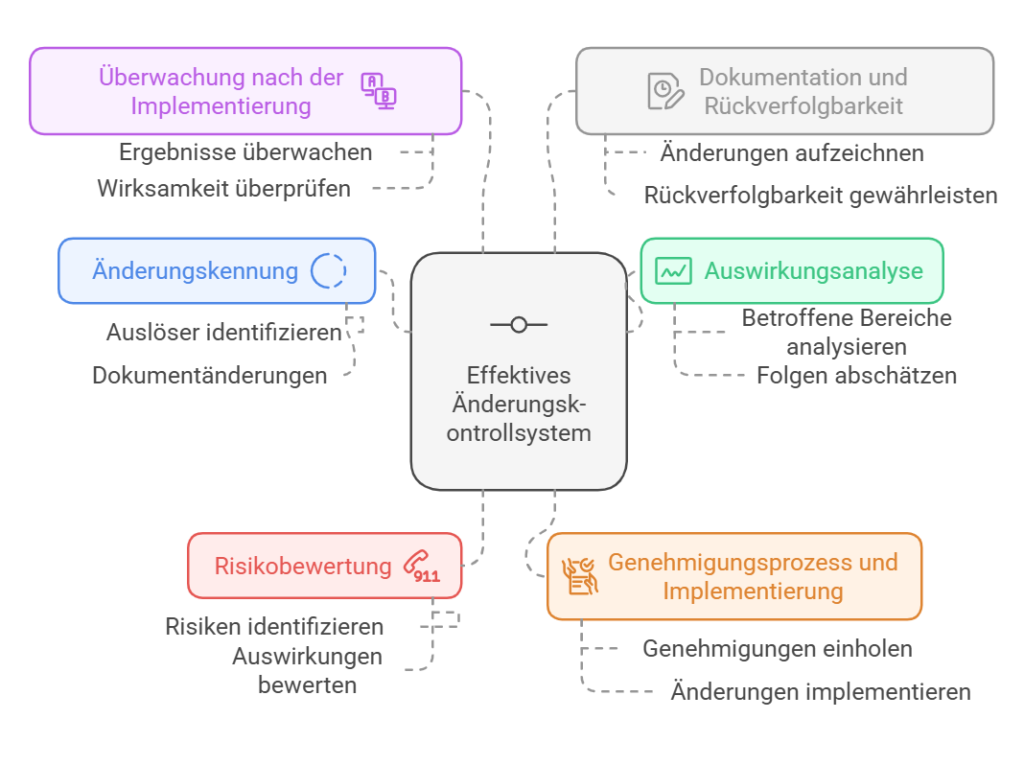
Die Auswirkungen bewerten: Beurteilen Sie, wie sich die vorgeschlagene Änderung auf die Funktion, Leistung und Sicherheit des Produkts auswirken könnte.
-
Risikobewertungen durchführen: Führen Sie gründliche Risikoanalysen durch, um potenzielle Risiken im Zusammenhang mit der Änderung zu identifizieren und zu mindern.
-
Dokumentation aktualisieren: Ändern Sie die Medizinprodukteakte (Device Master Record – DMR) und die Stückliste (Bill of Materials – BOM), um die Änderungen genau abzubilden.
-
Fertigungspartner einbeziehen: Binden Sie interne Fertigungsstätten und externe Lieferanten frühzeitig in den Design- und Entwicklungsprozess ein.
Signifikante Änderungen sind solche, welche die Sicherheit oder Wirksamkeit des Produkts beeinträchtigen oder zu wesentlichen Änderungen der Zweckbestimmung führen könnten.
Anpassungen der Qualitätskontrolle
Qualitätskontrollprozesse spielen eine entscheidende Rolle bei der Gewährleistung der Konsistenz und Zuverlässigkeit von Medizinprodukten. Bei der Implementierung von Änderungen an Qualitätskontrollverfahren sollten Hersteller:
-
Die Auswirkungen auf die Produktqualität bewerten: Beurteilen Sie, wie die vorgeschlagenen Änderungen die Gesamtqualität des Produkts beeinflussen könnten.
-
Prüfkriterien aktualisieren: Modifizieren Sie Kontrollpunkte der Qualitätssicherung und kritische Merkmale, um eine gründliche und sorgfältige Fehlererkennung aufrechtzuerhalten.
-
Änderungen validieren: Führen Sie notwendige Validierungsaktivitäten durch, um die Wirksamkeit der neuen Qualitätskontrollmaßnahmen sicherzustellen.
-
Personal schulen: Bieten Sie den an den Qualitätskontrollprozessen beteiligten Mitarbeitern umfassende Schulungen an, um eine ordnungsgemäße Umsetzung der Änderungen zu gewährleisten.
Best Practice: Konzentrieren Sie sich bei der Implementierung von Anpassungen der Qualitätskontrolle auf praktische Schritte zur nahtlosen Integration. Beginnen Sie mit einem Testlauf (Pilotlauf) der überarbeiteten Prozesse in kleinem Maßstab, um Probleme vor der vollständigen Implementierung zu identifizieren und zu lösen. Beziehen Sie funktionsübergreifende Teams ein, um Erkenntnisse zu gewinnen und sicherzustellen, dass alle Perspektiven berücksichtigt werden. Werkzeuge wie die FMEA (Fehlermöglichkeits- und Einflussanalyse) können verwendet werden, um die Auswirkungen von Änderungen systematisch zu bewerten. Überprüfen Sie nach der Implementierung regelmäßig die Fehlerquoten und Qualitätskennzahlen, um Verbesserungen zu bestätigen, und richten Sie eine Feedbackschleife zur kontinuierlichen Optimierung ein.
Modifikationen an der Ausrüstung
Änderungen an der Fertigungsausrüstung können erhebliche Auswirkungen auf die Produktqualität und -konsistenz haben. Bei der Modifikation von Ausrüstung sollten Hersteller:
-
Die Auswirkungen bewerten: Beurteilen Sie, wie sich Änderungen an der Ausrüstung auf den Herstellungsprozess und das Endprodukt auswirken könnten.
-
Risikobewertungen durchführen: Führen Sie eine gründliche Risikoanalyse gemäß Richtlinien wie der ISO 14971 durch, wobei Gefährdungen, vorhersehbare Ereignisse, Gefährdungssituationen und potenzielle Schäden berücksichtigt werden.
-
Kontrollen implementieren: Legen Sie angemessene Kontrollen fest, um die Produktsicherheit und -qualität nach der Implementierung der Ausrüstungsänderungen zu gewährleisten.
-
Den Prozess validieren: Bestimmen Sie basierend auf den Ergebnissen der Risikobewertung, ob eine Validierung des Herstellungsprozesses erforderlich ist.
Um Prozessänderungen in diesen Bereichen effektiv zu verwalten, sollten Hersteller ein formelles Änderungskontrollsystem implementieren.

Dieses formelle Änderungskontrollsystem sollte Folgendes umfassen:
-
Dokumentation von Änderungsanträgen: Verwenden Sie ein standardisiertes Formular, um vorgeschlagene Änderungen, betroffene Dokumente und Genehmigungsunterschriften zu erfassen.
-
Prüfprozess: Setzen Sie ein funktionsübergreifendes Team ein, um Änderungsanträge basierend auf ihrem Nutzen und ihren potenziellen Auswirkungen auf das QMS zu bewerten.
-
Implementierungsplanung: Entwickeln Sie detaillierte Pläne für die Ausführung genehmigter Änderungen, einschließlich Zeitplänen und Ressourcenallokation.
-
Aktualisierung der Dokumentation: Führen Sie umfassende Aufzeichnungen über alle änderungsbezogenen Aktivitäten, einschließlich Designprüfungen, Risikobewertungen, Risikominderungen und Validierungsergebnissen.
-
Schulung: Bieten Sie Mitarbeitern und Führungskräften fortlaufende Schulungen zum Änderungskontrollsystem und dessen Bedeutung an.
Durch die Implementierung des Änderungskontrollmanagements in ISO 13485-basierten Systemen können Medizintechnikhersteller sicherstellen, dass Modifikationen an Herstellungsprozessen, Qualitätskontrollverfahren und Ausrüstung kontrolliert und koordiniert eingeführt werden. Dieser Ansatz hilft, Risiken zu minimieren, die Produktqualität aufrechtzuerhalten und die Konformität mit regulatorischen Anforderungen nachzuweisen.
Implementing Change Control Management in ISO 13485: Verwaltung von Designänderungen
Designänderungen (Entwicklungsänderungen) sind ein integraler Bestandteil des Entwicklungsprozesses für Medizinprodukte. Sie können in jeder Phase auftreten, von der Phase vor der Markteinführung bis zur Post-Market-Phase, und müssen gemäß etablierten Verfahren verwaltet werden. Der Schlüssel zur effektiven Kontrolle von Designänderungen ist deren Integration in den Prozess der Entwicklungssteuerung, die Durchführung gründlicher Verifizierungen und Validierungen sowie die Aufrechterhaltung einer aktuellen Entwicklungsakte (Design History File – DHF).
Integration in die Entwicklungssteuerung
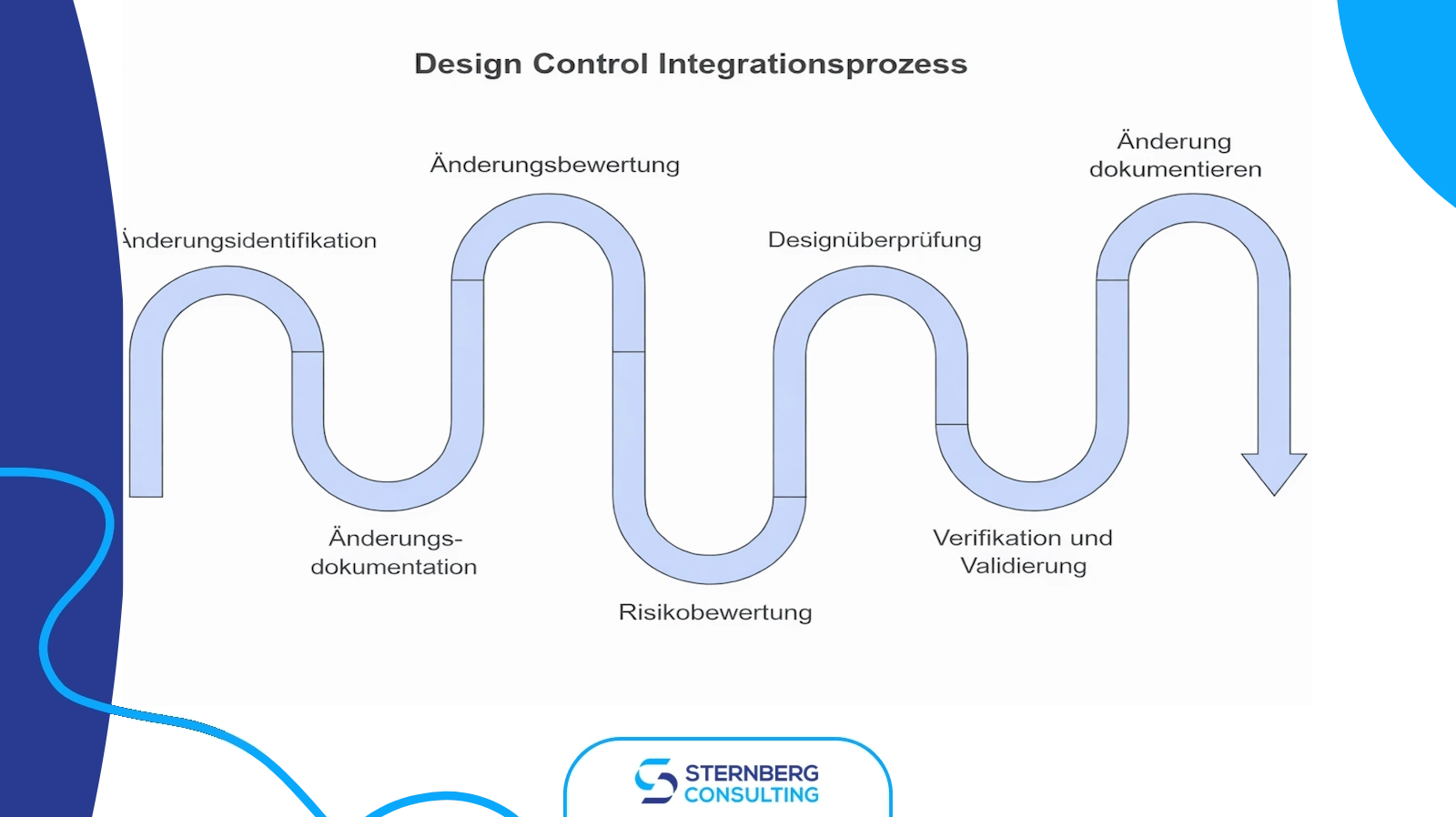
Die Integration von Designänderungen in den Prozess der Entwicklungssteuerung umfasst mehrere Schritte:
-
Identifizierung der Änderung: Erkennen Sie die Notwendigkeit einer Änderung, die aus verschiedenen Quellen resultieren kann, wie etwa Produktreklamationen, Audit-Feststellungen oder Initiativen zur fortlaufenden Verbesserung.
-
Dokumentation der Änderung: Halten Sie die Änderung in einem gelenkten Dokument fest, beispielsweise in einem Formular für einen Design-Änderungsantrag (Design Change Request – DCR). Fügen Sie eine klare Beschreibung, die Begründung, Ziele und alle unterstützenden Daten im Zusammenhang mit der vorgeschlagenen Änderung bei.
-
Bewertung der Änderung: Bewerten Sie die Auswirkungen der vorgeschlagenen Änderung auf die Funktion, Leistung, Gebrauchstauglichkeit und Sicherheit des Produkts. Bewerten Sie auch potenzielle Auswirkungen auf die Fertigung, Lieferanten, Qualitätskontrollen und regulatorische Einreichungen. Diese Bewertung sollte ein funktionsübergreifendes Team einbeziehen, um alle Aspekte der Änderung zu berücksichtigen.
-
Risikobewertung: Führen Sie eine gründliche Risikoanalyse (gemäß ISO 14971) durch, um potenzielle Risiken im Zusammenhang mit der Änderung zu identifizieren und zu mindern. Aktualisieren Sie bei Bedarf die Risikomanagement-Akte.
-
Design-Review (Entwicklungsprüfung): Führen Sie eine formelle Design-Prüfung durch, um Erkenntnisse darüber zu gewinnen, ob zusätzliche Verifizierungen oder Validierungen des Designs erforderlich sind, bevor die Änderung implementiert wird. Stellen Sie sicher, dass die Änderungen mit den Qualitäts- und regulatorischen Anforderungen übereinstimmen.
-
Verifizierung und Validierung: Verifizieren Sie, dass die Designänderung die spezifizierten Design-Inputs erfüllt und keine unbeabsichtigten Probleme verursacht. Validieren Sie die Änderung, um zu bestätigen, dass sie in der vorgesehenen Anwendungsumgebung effektiv funktioniert. Dokumentieren Sie die V&V-Ergebnisse als Teil der Entwicklungsaufzeichnungen.
-
Aufzeichnung der Änderung: Führen Sie umfassende Aufzeichnungen über alle mit der Änderung verbundenen Entwicklungsaktivitäten, einschließlich Design-Reviews, Inputs, Outputs und Risikobewertungen. Sie sollten auch Verifizierungs- und Validierungsdaten aufzeichnen und den Design-Änderungsantrag sowie die Entwicklungsakte (DHF) aktualisieren, um die Rückverfolgbarkeit zu gewährleisten.
Verifizierung und Validierung
Verifizierung und Validierung sind entscheidende Schritte im Designänderungsprozess, die sicherstellen, dass das modifizierte Produkt die spezifizierten Anforderungen und die Bedürfnisse der Anwender erfüllt.
Verifizierung:
-
Bestätigt, dass die Design-Outputs die Design-Inputs erfüllen.
-
Beinhaltet Tests zum Nachweis, dass die Zweckbestimmung des Produkts durch Befolgen dokumentierter Verfahren präzise umgesetzt wird.
-
Ist für jede Änderung obligatorisch, unabhängig von deren Umfang.

Validierung:
-
Belegt, dass das Produkt die Bedürfnisse der Anwender erfüllt und unter den erwarteten Nutzungsbedingungen zuverlässig funktioniert.
-
Ist bei geringfügigen Änderungen möglicherweise nicht immer erforderlich.
-
Ist erforderlich, wenn die Änderung beeinflusst, wie das Produkt definierte Anwenderbedürfnisse erfüllt.
Hinweis: Die Verifizierung kann erst erfolgen, wenn Design-Outputs und -Inputs abgeschlossen sind. Die Validierung kann erst stattfinden, wenn die Anwenderbedürfnisse definiert sind.
Aktualisierung der Entwicklungsakte (Design History File)
Die Entwicklungsakte (DHF) ist eine umfassende Aufzeichnung der Entwicklung eines Produkts über seinen gesamten Lebenszyklus hinweg. Die ordnungsgemäße Verwaltung und Aktualisierung der DHF ist für die Aufrechterhaltung der regulatorischen Compliance und den Nachweis der Einhaltung von Qualitätsstandards unerlässlich.
Wichtige Aspekte bei der Aktualisierung der DHF umfassen:
-
Kontinuierliche Dokumentation: Entwickler müssen die DHF während des gesamten Lebenszyklus eines Produkts kontinuierlich aktualisieren und Designänderungen präzise erfassen, um ein Höchstmaß an Patientensicherheit und Produktqualität zu gewährleisten.
-
Rückverfolgbarkeit: Stellen Sie sicher, dass alle Änderungen rückverfolgbar und zugänglich sind; sie dienen als Beweis für die gebotene Sorgfalt und das Engagement für Qualität und Sicherheit.
-
Dokumentation der Änderungskontrolle: Integrieren Sie die Dokumentation der Änderungskontrolle, um Modifikationen und Aktualisierungen über den gesamten Lebenszyklus des Produkts zu verfolgen.
-
Elektronisches Management: Nutzen Sie fortschrittliche Dokumentenmanagementsysteme mit robusten Funktionen zur Versionskontrolle und für elektronische Signaturen, um die Integrität und Sicherheit der DHF zu gewährleisten.
-
Periodische Überprüfungen: Führen Sie regelmäßige Audits und Prüfungen durch, um die Relevanz der DHF und die Konformität mit sich entwickelnden regulatorischen Anforderungen aufrechtzuerhalten.

Durch das effektive Management von Designänderungen mittels ordnungsgemäßer Integration in die Entwicklungssteuerung, gründlicher Verifizierung und Validierung sowie gewissenhafter Aktualisierung der Entwicklungsakte können Medizintechnikhersteller die laufende Konformität mit der ISO 13485 und den FDA-Vorschriften sicherstellen und gleichzeitig die Produktqualität und -sicherheit wahren.
Änderungskontrolle bei Lieferanten
Die Änderungskontrolle bei Lieferanten ist ein wesentlicher Aspekt des Qualitätsmanagements in der Medizintechnikbranche. Sie stellt sicher, dass Modifikationen seitens der Lieferanten nicht die Sicherheit, Wirksamkeit oder regulatorische Konformität des fertigen Produkts beeinträchtigen. Hersteller von Medizinprodukten müssen robuste Prozesse etablieren, um Lieferantenänderungen effektiv zu verwalten.
Lieferantenvereinbarungen (Qualitätssicherungsvereinbarungen)
Lieferantenvereinbarungen bilden das Fundament einer effektiven Änderungskontrolle bei Lieferanten. Diese Verträge sollten die Verantwortlichkeiten und Erwartungen beider Parteien klar umreißen.

Wichtige Elemente, die in Lieferantenvereinbarungen enthalten sein sollten:
-
Qualitätsanforderungen
-
Kommunikationsprotokolle
-
Verfahren zur Änderungsmitteilung
-
Meldung von Nichtkonformitäten
Es ist entscheidend festzulegen, dass Lieferanten das Medizintechnikunternehmen über alle geplanten Änderungen an Produkten oder Prozessen informieren müssen. Diese Anforderung sollte explizit in der Qualitätssicherungsvereinbarung (QSV) festgehalten werden. Auf diese Weise können Hersteller die Kontrolle über den gesamten Outsourcing-Prozess behalten und die Einhaltung regulatorischer Standards sicherstellen.
Bewertung von Lieferantenänderungen
Wenn ein Lieferant eine Änderung vorschlägt, müssen Medizintechnikhersteller eine gründliche Auswirkungsanalyse durchführen. Dieser Bewertungsprozess umfasst typischerweise folgende Schritte:
-
Prüfung der Änderungsmitteilung: Analysieren Sie die vorgeschlagene Änderung und deren potenzielle Auswirkungen auf das Produkt oder den Prozess.
-
Risikobewertung: Führen Sie eine umfassende Risikoanalyse durch, um potenzielle Gefährdungen und deren Folgen zu identifizieren.
-
Auswirkungsanalyse (Impact Analysis): Beurteilen Sie, wie die Änderung die Form, Passgenauigkeit und Funktion (Form, Fit, and Function – FFF) einzelner Teile sowie deren Gesamtleistung im fertigen Produkt beeinflussen könnte.
-
Due Diligence: Stellen Sie sicher, dass neu gefertigte Teile äquivalent zu den ersetzten Teilen sind, insbesondere bei einem Lieferantenwechsel.
-
Regulatorische Bewertung: Bestimmen Sie, ob die Änderung zusätzliche regulatorische Einreichungen oder Genehmigungen erfordert.
Um diesen Bewertungsprozess zu erleichtern, können Hersteller Scorecards zur Lieferantenbewertung nutzen. Dieses Werkzeug hilft dabei, die Leistung der Lieferanten in verschiedenen Kategorien zu quantifizieren, zu verfolgen und zu verwalten, einschließlich Produktqualität, Kundenservice, Kosten und Lieferung.
Auswirkungen auf das fertige Produkt
Die Auswirkungen von Lieferantenänderungen auf das fertige Produkt können erheblich und weitreichend sein.
Hersteller müssen sowohl unmittelbare als auch langfristige Effekte berücksichtigen, einschließlich:
-
Produktqualität: Beurteilen Sie, wie die Änderung die Gesamtqualität und Leistung des fertigen Produkts beeinflussen könnte.
-
Regulatorische Compliance: Bewerten Sie, ob die Änderung mit bestehenden regulatorischen Zulassungen und Anforderungen übereinstimmt.
-
Herstellungsprozesse: Bestimmen Sie, ob die Änderung Modifikationen an internen Fertigungs- oder Montageprozessen erforderlich macht.
-
Lieferkette: Berücksichtigen Sie die potenziellen Auswirkungen auf Lagerbestände, Durchlaufzeiten und die allgemeine Effizienz der Lieferkette.
-
Kundenzufriedenheit: Beurteilen Sie, wie die Änderung die Endnutzererfahrung und die Kundenerwartungen beeinflussen könnte.
Es ist wichtig zu beachten, dass selbst kleine Änderungen erhebliche Auswirkungen haben können. Beispielsweise kann der Wechsel von einem Lieferanten zu einem anderen bei einer kritischen Komponente Geschäftsrisiken bergen und umfangreiche Qualifizierungsprozesse erfordern.
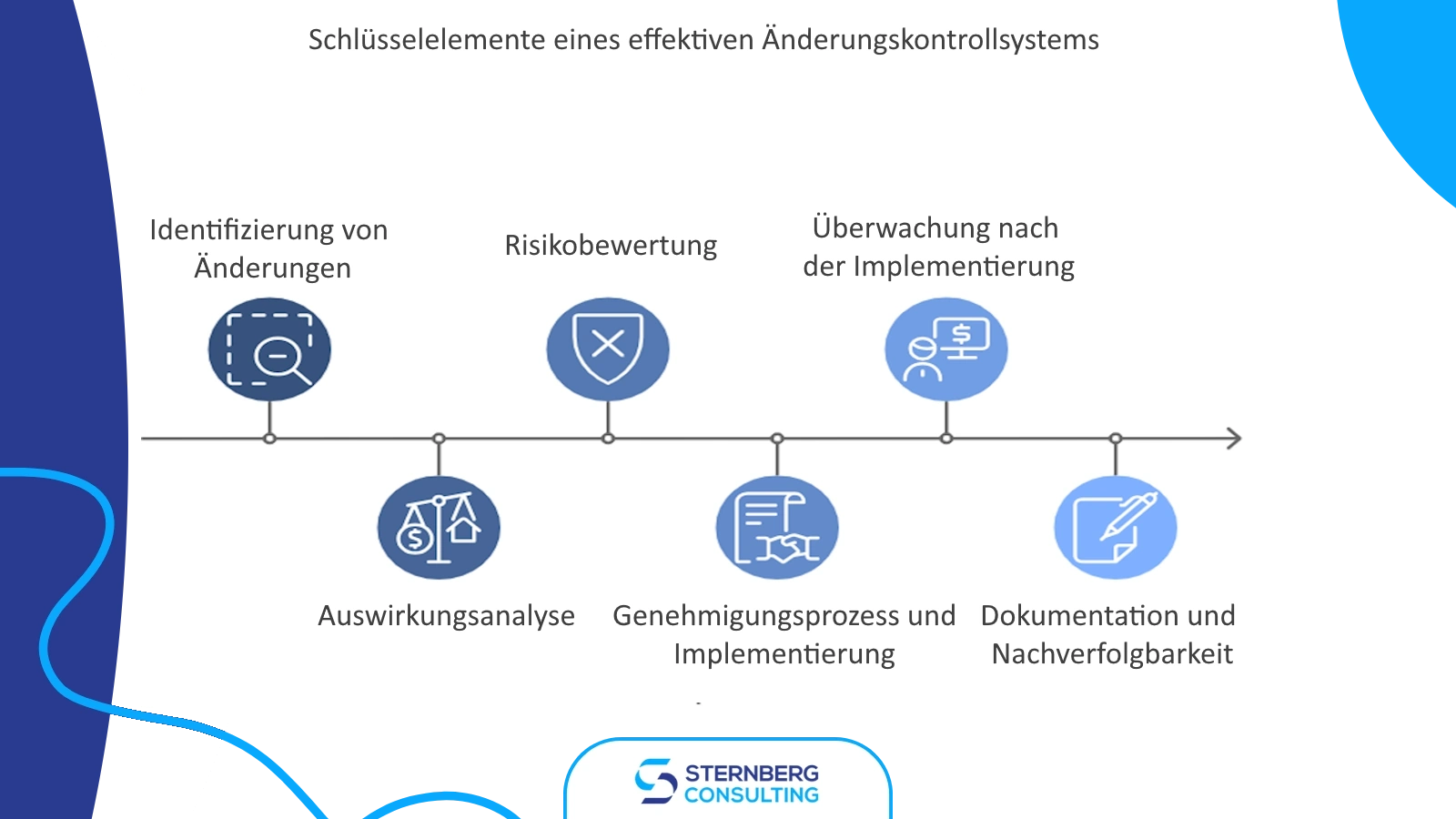
Um diese Auswirkungen effektiv zu verwalten, sollten Hersteller:
-
Ein formelles Änderungskontrollsystem mit standardisierter Dokumentation und Genehmigungsprozessen implementieren.
-
Gründliche Bewertungen vor der Implementierung jeder Änderung durchführen, unabhängig von deren wahrgenommenem Umfang.
-
Umfassende Aufzeichnungen über alle änderungsbezogenen Aktivitäten führen, einschließlich Bewertungen, Risikobanalysen, Risikominderungen und Validierungsergebnissen.
-
Mitarbeiter fortlaufend über die Bedeutung der Änderungskontrolle bei Lieferanten und deren Auswirkungen auf Produktqualität und Compliance schulen.
Durch die Implementierung robuster Prozesse zur Änderungskontrolle bei Lieferanten können Medizintechnikhersteller die Produktqualität aufrechterhalten, die regulatorische Konformität sicherstellen und Risiken im Zusammenhang mit Modifikationen durch Lieferanten minimieren. Dieser Ansatz steht im Einklang mit den Anforderungen von FDA 21 CFR 820.50 und ISO 13485:2016 Abschnitt 7.4 und demonstriert ein Bekenntnis zu Exzellenz im Qualitätsmanagement und in der Zusammenarbeit mit Lieferanten.
Fazit
Die Implementierung des Änderungskontrollmanagements gemäß ISO 13485:2016 bedeutet den Aufbau eines umfassenden Prozesses mit detaillierter Dokumentation, gründlichen Risikobewertungen, effektiver Kommunikation und funktionsübergreifender Zusammenarbeit. Diese Elemente gewährleisten die erfolgreiche Umsetzung von Änderungen und stärken die Fähigkeit einer Organisation, zu innovieren und auf Herausforderungen zu reagieren.
Letztendlich fördert ein gut implementiertes Änderungskontrollsystem das Vertrauen der Stakeholder, sichert die Produktkonsistenz und steht im Einklang mit globalen regulatorischen Standards. Durch die Priorisierung eines strukturierten Änderungsmanagements können Medizintechnikhersteller die Qualität und Leistung ihrer Produkte sichern, die Patientensicherheit erhöhen und das Vertrauen in ihre Marke nachhaltig stärken.
Sind Sie bereit für die Implementierung der Änderungskontrolle? Kontaktieren Sie uns jetzt und lassen Sie uns gemeinsam daran arbeiten, Ihre Ziele für die ISO-Zertifizierung zu erreichen.
Häufig gestellte Fragen (FAQs)
Warum ist die Änderungskontrolle für Medizintechnikunternehmen so wichtig?
Die Änderungskontrolle stellt sicher, dass Modifikationen an Managementprozessen, Designs oder Systemen die Produktqualität, Sicherheit oder regulatorische Konformität nicht beeinträchtigen. Sie hilft dabei, die Rückverfolgbarkeit zu wahren, stellt die Verantwortlichkeit der Stakeholder sicher und ermöglicht eine effektive Risikominderung, um unbeabsichtigte Folgen von Änderungen anzugehen.
Welche Arten von Änderungen erfordern eine formelle Änderungskontrolle gemäß ISO 13485?
Änderungen, die das Design, den Herstellungsprozess, die Lieferantenqualifikationen, regulatorische Einreichungen oder die Produktkennzeichnung betreffen, erfordern in der Regel eine formelle Änderungskontrolle. Jede Modifikation, welche die Produktleistung, die Gebrauchstauglichkeit oder die Compliance beeinflussen könnte, muss systematisch bewertet und verwaltet werden.
Welche Dokumentation ist für einen effektiven Änderungskontrollprozess erforderlich?
Eine effektive Änderungskontrolle erfordert eine umfassende Dokumentation von:
-
Änderungsanträgen, einschließlich Begründung und Umfang.
-
Risikobewertungen und Auswirkungsanalysen.
-
Design-Reviews und Validierungsaktivitäten.
-
Genehmigungen durch relevante Stakeholder.
-
Aktualisierungen von Verfahren, Schulungsmaterialien und regulatorischen Einreichungen.
Wie sollte die Schulung im Kontext der Änderungskontrolle verwaltet werden?
Schulungen sind entscheidend, um sicherzustellen, dass das Personal Änderungen versteht und korrekt umsetzt. Schulungen sollten nach der Genehmigung, aber vor der Implementierung durchgeführt werden und Aktualisierungen von Verfahren, Workflows und Rollen umfassen. Schulungsaufzeichnungen müssen für Audit-Zwecke aufbewahrt werden.
Wie kann Technologie die ISO 13485-Änderungskontrollprozesse unterstützen?
Digitale Werkzeuge und Softwaresysteme können die Änderungskontrolle optimieren, indem sie:
-
Die Versionskontrolle von Dokumenten und Genehmigungs-Workflows automatisieren.
-
Echtzeit-Transparenz über den Status von Änderungen bieten.
-
Die Zusammenarbeit zwischen funktionsübergreifenden Teams erleichtern.
-
Rückverfolgbarkeit und Audit-Bereitschaft durch zentralisierte Aktenführung sicherstellen.